MTD
Mammalian Transcriptomic Database
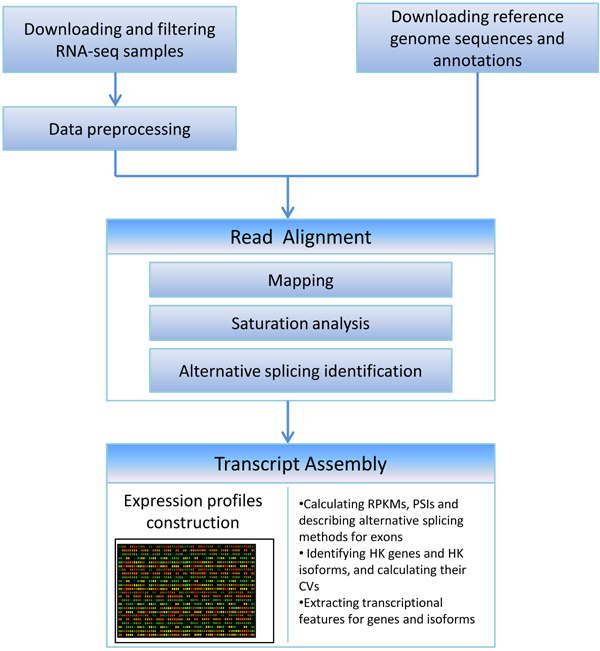
Pipeline
- Filtering raw RNA-seq samples and preprocessing
a. All raw RNA-seq data were chosen from healthy samples without any special treatments in the public database SRA. Five criteria are considered as follows:
(1) Only selecting samples under normal conditions and removing tissues/cell lines with special treatments to avoid different regulation mechanisms under diverse physiological conditions.
(2) Removing all mixed tissue/cell line samples that might mask the real differences among samples.
(3) Filtering severe unsaturated datasets that might increase the false negative rate of gene expression and the HK gene.
(4) Selecting the most saturated and highest sequencing quality samples for each tissue/cell line.
(5) Selecting samples from the Illumina Genome Analyzer and Illumina HiSeq 2000/2500 to reduce the differences resulting from the use of different sequencing instruments.
b. After manual filtering, data preprocessing is conducted with six criteria as follows:
(1) Filtering out adaptor reads.
(2) Truncating reads with more than 2% 'N' bases.
(3) Truncating reads with low quality (below 20) over 20% of the length.
(4) Truncating reads with low quality (below 13) over 10% of the length.
(5) Truncating reads with a sequence quality of less than 20.
(6) After truncating, any human and mouse reads shorter than 50 bp (36 bp for rat and pig because of the poor quality of the relevant RNA-seq resources) will be filtered out.
- Read mapping, transcript assembly and detecting alternative splicing events by Tophat and Cufflinks
- Saturation analysis by Bedtools
- Identification of housekeeping genes and isoforms (RPKM >0 in all the representative experiments of the tissues/cell lines with saturated data)